Vad du behöver veta om sickle cell sjukdom
Termen sickle cell disease (SCD) innehåller flera ärftliga abnormiteter i röda blodkroppar. SCD-patienter har onormalt hemoglobin som kallas hemoglobin S eller sicklehemoglobin i sina röda blodkroppar.
Hemoglobin är ett protein i röda blodkroppar som bär syre i kroppen.
Uttrycket "arv" indikerar att denna sjukdom orsakas av gener som släppts av föräldrar till sina barn. Till skillnad från förkylning eller infektion är SCD inte smittsam så att friska människor inte kommer att utsättas för SDC från drabbade.
SCD lider arv två onormala hemoglobingener från far och mor. Vilken typ av SCD som helst får åtminstone en av de två onormala generen att kroppen producerar hemoglobin S. Under tiden kommer någon som har två hemoglobin S-gener (eller SS-hemoglobin) att lida av en sjukdom som kallas sickle cellanemi. Sickle cellanemi är den mest allvarliga och vanligaste typen av SCD.
SC-hemoglobin och hemoglobin S-thalassemi är två andra vanliga former av SCD.
Några former av sicklecell sjukdom:
- Hemoglobin SS
- Hemoglobin SC
- Sp0-hemoglobin-thalassemi
- Hemoglobin Sp + talassemi
- Hemoglobin SD
- Hemoglobin SE
Hur sicklecell sjukdom kan uppstå?
Cellens vävnader behöver en syreförsörjning för att fungera korrekt. Vanligtvis tar hemoglobin i röda blodkroppar syre från lungorna och bär den till alla kroppsvävnader.
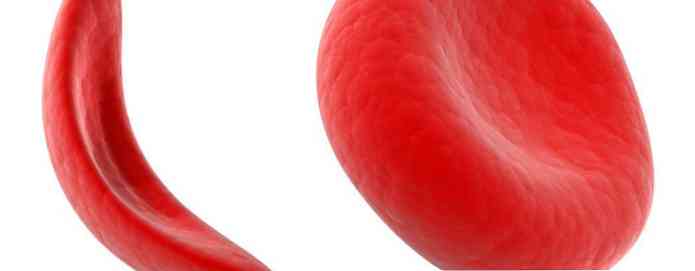
Röda blodkroppar som innehåller normalt hemoglobin är formade som bitar (som munkar utan hål). Denna form låter cellerna bli flexibla så att de kan flytta genom stora och små blodkärl för att bära syre.
Sicklehemoglobin är inte som normalt hemoglobin. Sicklehemoglobin bildar en styv stam i röda blodkroppar, blir till en segel eller är segel.
Crescent celler är oflexibla och kan hålla fast vid kärlväggen, vilket orsakar blockeringar som saktar eller stoppar blodflödet. När detta tillstånd uppstår, kan syre inte nå den omgivande vävnaden.
Brist på vävnad syre kan orsaka allvarlig smärta som kallas smärta kriser. Denna smärta kan plötsligt träffas utan varning så att patienterna behöver effektiv behandling på sjukhuset.
De flesta barn med SCD upplever inga smärta eller smärta kriser, medan ungdomar och vuxna kan lida av kronisk, pågående smärta.
Onormala röda blodkroppar och lågt syreintag kan också orsaka organskador, som mjälte, hjärna, ögon, lungor, lever, hjärta, njurar, penis, leder, hud eller till och med ben.
Eftersom formen inte är flexibel kan siglceller explodera eller kallas hemolyze. Den normala livscykeln för röda blodkroppar varierar från 90 till 120 dagar, medan sicklecellerna endast kan varas i 10 till 20 dagar.
Kroppen gör alltid nya röda blodkroppar för att ersätta gamla celler. Emellertid har kroppen av SCD-patienter svårt att balansera ny blodcellsproduktion med snabb destruktion av blodkroppar. Som ett resultat tenderar antalet röda blodkroppar att vara lägre än det normala antalet. Detta tillstånd kallas anemi som en orsak till kroppens reducerade energi.
Huruvida sicklecellanemi kan botas?
Sjukcellssjukdom är en livslång sjukdom. Svårighetsgraden av sjukdomen hos varje patient kommer att variera.
I höginkomstländer som Förenta staterna är livslängden hos SCD-patienter för närvarande cirka 40-60 år. 1973 var medelåldern för SCD-patienter i Förenta staterna endast 14 år. Detta visar framsteg vid diagnos och behandling av SCD.
För närvarande är hematopoetisk stamcellstransplantation (HSCT) det enda botemedlet för SCD. Tyvärr är de flesta SCD-lider för gamla för att genomgå en transplantation eller inte ha släktingar som har en genetisk matchning som en givare. Lämpliga donatorer behövs för att få de bästa transplantationsresultaten.
Det finns många effektiva behandlingar som kan minska symtomen och förlänga livslängden hos SDC-patienter. Tidig diagnos och vanlig sjukvård för att förhindra komplikationer bidrar också till att förbättra livskvaliteten för patienter.